

The date February 2, 2026, was not just another deadline on the regulatory calendar; it marked the most significant shift in U.S. medical device oversight in three decades. With the final implementation of the Quality Management System Regulation (QMSR), the FDA officially retired the legacy Quality System Regulation (QSR) in favor of a framework harmonized with ISO 13485:2016.
For C-suite executives and MedTech innovators, this transition represents a fundamental change in how “quality” is measured. No longer can the Quality Management System (QMS) be viewed as a static archive of documents. Instead, it is now an active, risk-based engine of business continuity. The Medical Device Single Audit Program (MDSAP), often misunderstood as a mere administrative convenience, has emerged as the premier strategic tool for navigating this new landscape.
The Strategic Fallacy: Why ‘Compliance as a Commodity’ Fails Executives
A common misconception among healthcare leadership is that regulatory compliance is a “tax on innovation”—a commodity to be managed at the lowest possible cost. This “check-the-box” mentality often leads organizations to treat MDSAP as nothing more than “ISO 13485 with extra country-specific steps.”
This perspective is a strategic fallacy. Under the QMSR, the FDA has moved away from the Quality System Inspection Technique (QSIT) and adopted a more holistic approach (Compliance Program 7382.850) that scrutinizes process interactions and risk-based decision-making throughout the product lifecycle.
When executives treat compliance as a commodity, they miss the de-risking potential of the MDSAP framework. A fragmented QMS—where one team handles FDA 21 CFR 820 and another manages ISO 13485 for the European or Canadian markets—creates silos of data that are prone to contradictions. These contradictions are “red meat” for modern auditors. Under QMSR, previously exempt records such as management reviews, internal audits, and supplier audit reports are now subject to FDA inspection.
If your organization is participating in MDSAP, you are already operating under a high-transparency model. Executives who leverage MDSAP as a centralized “source of truth” rather than a series of disparate audits gain a competitive edge. They reduce “audit fatigue,” ensure a unified global quality culture, and, most importantly, provide the board with a quantified view of regulatory risk across five major jurisdictions: Australia, Brazil, Canada, Japan, and the United States.
Evidence-Based Readiness: Mapping MDSAP to the New FDA Landscape
The synergy between MDSAP and the 2026 QMSR enforcement is not accidental; it is by design. The FDA’s incorporation of ISO 13485:2016 by reference means that the foundational requirements for the U.S. market now mirror the core of the MDSAP audit model.
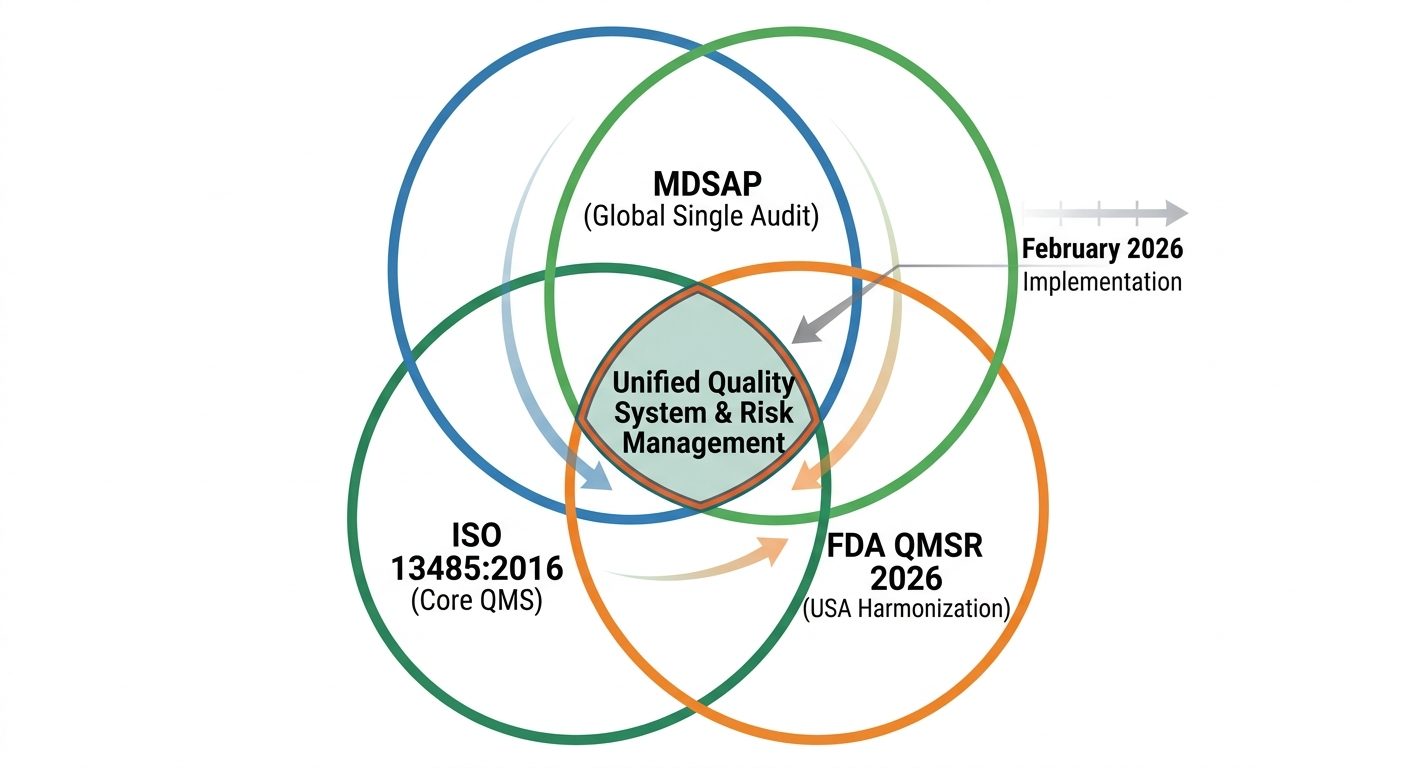
1. Terminology Harmonization and the “Medical Device File”
One of the most visible changes in the QMSR is the shift in terminology. The legacy “Device Master Record (DMR)” has been supplanted by the ISO-aligned Medical Device File (MDF). While the intent remains similar—ensuring the specifications and procedures for a device are documented—the MDF requires a more integrated approach to technical documentation. MDSAP participants are already accustomed to the MDF structure, which eases the burden of transitioning legacy U.S. records to the new standard.
2. Risk Management Across the Lifecycle
Under the old QSR, risk management was primarily focused on design validation. The QMSR (and ISO 13485) elevates risk-based thinking to every facet of the QMS, from purchasing and supplier controls to post-market surveillance.
Industry experts, such as the consultants at Operon Strategist, emphasize that a successful MDSAP transition requires a deep gap analysis that moves beyond documentation into active process-based evidence. This involves mapping how a single risk identified in a complaint is fed back into the design control process and reflected in updated clinical evidence—a “closed-loop” system that is the hallmark of a mature MDSAP-certified organization.
3. Supplier Controls and Technical Traceability
The MDSAP audit model places significant weight on Purchasing and Supplier Controls. With the 2026 QMSR, the FDA has signaled a more rigorous expectation for documented supplier qualification. Organizations that have utilized MDSAP to harmonize their supplier audits across global markets find themselves ahead of the curve, as they already possess the “evidence of control” that QMSR inspectors now expect as a baseline.
Quantifying Success: The Evidence-Driven Approach
To de-risk innovation, executives must demand data. The MDSAP Nonconformity Grading System provides a quantified metric for QMS health that is recognized by all five participating regulators. A grade 4 or 5 nonconformity in an MDSAP audit is a clear signal of systemic failure that requires immediate C-suite attention.
Evidence from the International Medical Device Regulators Forum (IMDRF) suggests that companies participating in MDSAP experience fewer individual inspections and a higher degree of predictability in their regulatory path (IMDRF, 2021). By using the MDSAP audit report as a primary management tool, leadership can move from reactive “firefighting” during inspections to proactive “operational discipline.”
As the MedTech industry settles into the post-QMSR era, the divide between leaders and laggards will be defined by their ability to integrate these global standards. For companies looking to expand into the Indian market or other emerging regions, a robust MDSAP-aligned QMS provides the “clinical and regulatory evidence” required to prove that their innovation is not just a theoretical idea, but a verified, safe, and effective healthcare solution.
References
- Food and Drug Administration. (2024). Quality Management System Regulation: Final Rule. 21 CFR Part 820.
- International Medical Device Regulators Forum. (2021). Medical Device Single Audit Program (MDSAP) Working Group: N11 – Assessment of Auditing Organizations. IMDRF/MDSAP WG/N11 FINAL:2021.
- International Organization for Standardization. (2016). ISO 13485:2016 – Medical devices — Quality management systems — Requirements for regulatory purposes.
- MDSAP Regulatory Authorities. (2026). MDSAP Audit Approach – Revised February 2026.
