

Bringing a medical device from concept to market calls for thoughtful planning, a strong grasp of regulatory requirements, and a well-executed strategy. Strict international regulations like the FDA in the US, the EU MDR in Europe, and ISO 13485 quality standards can make this process difficult for new and developing businesses. These rules protect product performance and patient safety, so they are more than just administrative difficulties. Clinical evidence consistently shows that devices developed within structured regulatory and quality frameworks are more likely to achieve positive patient outcomes and long-term market success. Early incorporation of regulatory requirements into product design, for example, greatly lowers post-market safety concerns and expensive recalls, according to a study published in the Journal of Medical Devices. This article will outline doable tactics that startups can employ to adhere to global regulations, overcome common roadblocks, and leverage compliance to spur innovation and obtain a competitive advantage.
Understanding Key Regulatory Pathways
- FDA (U.S.): FDA (U.S.): The FDA divides medical devices into three risk-based groups, known as Class I, II, and III. While Class III devices need Premarket Approval (PMA), which entails strong clinical evidence, Class II devices typically need a 510(k) premarket notification to show substantial equivalency to an existing device. Pre-submission meetings with the FDA can help startups by giving them feedback and expediting the approval process.
- EU MDR (Europe): The EU MDR requires every device to go through a conformity assessment before it is allowed to be marketed. The assessment includes device classification, clinical evaluation, and CE marking. The notified bodies play a key role in evaluating compliance, and the new companies should ensure that their documentation, together with quality systems, meets the MDR standards.
- Health Canada (Canada): Under the Medical Devices Regulations, Health Canada (Canada) oversees the regulation of medical devices. Based on risk, devices are divided into four classes (I–IV). Before marketing a product in Canada, manufacturers of Class II, III, and IV devices must obtain a Medical Device Licence (MDL), which certifies that they have complied with safety and efficacy regulations.
A Medical Device Establishment Licence (MDEL), which guarantees proper protocols for distribution and complaint handling, is also required of businesses engaged in the production, importation, or distribution of Class I devices. Higher-class devices must have a quality management system certified to MDSAP, and Health Canada regularly audits for continued compliance.
- TGA (Australia): The Therapeutic Goods Administration (TGA) oversees medical devices under the Therapeutic Goods Act. Devices are classified from Class I to Class III and Active Implantable Medical Devices (AIMD) based on risk. Before any device can be supplied in Australia, it must be listed in the Australian Register of Therapeutic Goods (ARTG). Higher-risk device manufacturers must provide conformance assessment evidence, often supporting their applications with approvals from similar regulators (such as the FDA or EU). The TGA may conduct audits or request additional clinical and technical documentation to verify compliance with core safety and performance principles.
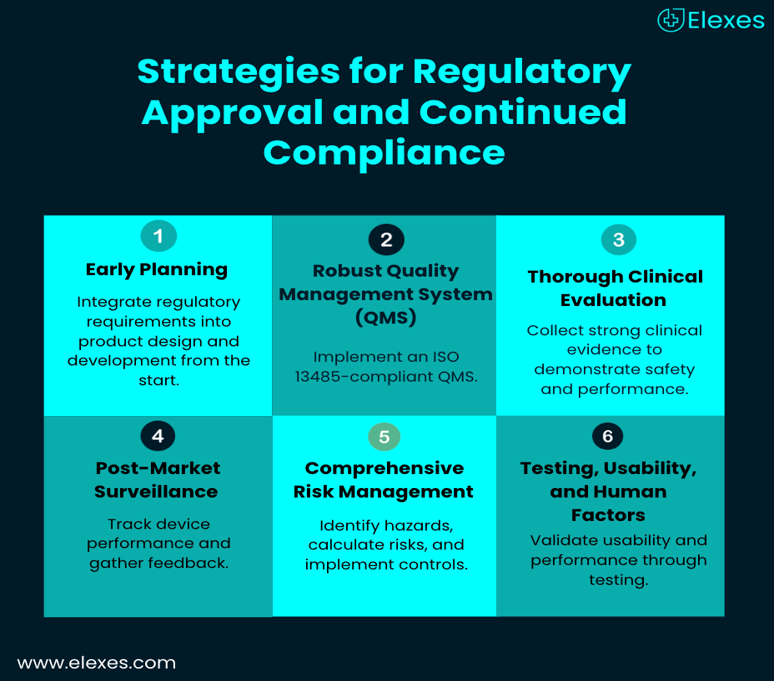
Strategies for Regulatory Approval and Continued Compliance
- Early Planning: Integrate regulatory requirements into product design and development from the start. Early planning helps identify potential compliance gaps and reduces the risk of costly redesigns or delays in approvals.
- Robust Quality Management System (QMS): A well-implemented ISO 13485-compliant QMS ensures that processes cover all aspects of regulatory compliance. This includes design verification and validation, document and change control, supplier management, and risk assessment.
- Thorough Clinical Evaluation: Thorough Clinical Evaluation: Collecting strong clinical evidence demonstrates a device’s safety and performance. Clinical evaluation is a requirement for higher-risk devices under both FDA and EU MDR regulations. Startups should plan clinical studies early to align with regulatory expectations. Sometimes, clinical evaluation requirements can be fulfilled through a literature review, while in other cases, a comprehensive clinical study is essential.
- Post-Market Surveillance: To track the performance of devices in the real world, post-market surveillance is crucial. This includes user feedback, adverse event reporting, and clinical follow-ups after the product is marketed. Adherence is maintained, and continuous product improvement is facilitated by a methodical monitoring system.
- Comprehensive Risk Management: It is crucial to put in place a structured risk management procedure that complies with ISO 14971. Safety problems can be avoided later by identifying possible device hazards, calculating risks, and putting in place efficient control measures early. To reduce lingering risks, risk controls can include protective measures, design elements, and unambiguous labelling. Throughout the device lifecycle, ongoing risk assessment guarantees that any new risks discovered after the product is put on the market are quickly fixed.
- Testing, Usability, and Human Factors: Thorough performance and usability testing validate that the device works as intended under real-world conditions. In order to ensure that the design supports safe and efficient use, human factors engineering detects possible use errors before market entry. Usability assessments for higher-risk devices are prioritised by regulatory agencies such as the FDA and EU MDR, which frequently call for human factors research to verify that crucial tasks can be completed accurately and safely.
- Global Submission Strategy: By putting in place a carefully considered global submission plan, businesses can better align product development with different regulatory frameworks. By mapping overlapping requirements across regions, including the FDA, EU MDR, Health Canada, and TGA, repetitive testing and paperwork can be minimised. Utilising standardised technical documentation and international standards (like the IMDRF guidelines) expedites submissions, speeds up approvals, and facilitates a quicker, more effective global market rollout.
Common Challenges and How to Overcome Them
- Resource Constraints: Startups usually do not have enough budget or other resources. The cost of hiring full-time employees can be avoided by outsourcing to experienced professionals to handle regulatory affairs and quality assurance.
- Complex Documentation: Regulatory submissions involve extensive documentation. Using an electronic quality management system (eQMS) can simplify document control, versioning, and compliance tracking, ensuring readiness for audits and inspections. Regulatory specialists or an expert can help by organising technical files, creating submission dossiers, and making sure that everything complies with regulations.
- Evolving Regulations: The approval timeline for devices could be extended due to the continuous changes in regulations. Startups should take part in training, subscribe to newsletters, and be active in the official regulatory channels so that they can quickly adapt to the new requirements. Compliance and regulatory professionals are capable of interpreting new guidelines, assessing their impact on existing practices, and successfully implementing necessary updates.
- Incorrect Classification or Pathway Selection: Inaccurate regulatory pathway selection or device classification can result in significant delays, rework, and higher expenses. Determining the appropriate classification and submission route requires early regulatory assessment. Engagement with experts from the very beginning leads to the right classification, a suitable regulatory approach, and swift approvals in different countries.

Leveraging Regulatory Approval and Compliance as a Competitive Advantage
Regulatory compliance requires legal obligation, and it can also be a strategic advantage for a startup. The early incorporation of standards such as FDA, EU MDR, and ISO 13485 into the product development process can put in place a number of things that will translate into competitive benefits for startups, thereby accelerating their growth and market entry.
- Building Stakeholder Trust: Compliance demonstrates that a device meets rigorous safety and performance standards. Investors, healthcare providers, and patients are more likely to trust startups with a proven regulatory track record. This trust can accelerate funding, partnerships, and market adoption.
- Enabling Global Market Expansion: Devices compliant with FDA, EU MDR, and ISO 13485 requirements are better positioned for international markets. Regulatory awareness lowers obstacles, expedites approval processes, and gives startups the confidence to grow internationally.
- Facilitating Reimbursement and Procurement: Health systems and insurers often require documented regulatory compliance before covering or procuring new devices. Startups with clear approvals and certifications can negotiate faster, smoother, and more favorable reimbursement arrangements.
- Minimizing Risk of Delays and Recalls: A proactive compliance approach reduces the likelihood of regulatory setbacks, product recalls, or penalties. Well-documented quality systems and adherence to standards ensure a smoother product lifecycle and fewer costly interruptions.
- Differentiating Products in a Competitive Market: In competitive digital health and medtech sectors, regulatory compliance can be a unique selling point. Demonstrating rigorous adherence to standards reassures providers, patients, and investors that the product is reliable and safe.
- Supporting Innovation Safely: Embedding regulatory requirements early in product design guides innovation without compromising safety. Startups can experiment with new technologies while staying compliant, transforming legal requirements into a framework for developing products safely and efficiently.
- Example in Practice: A startup making wearable cardiac monitors took advantage of ISO 13485 compliance and CE marking to enter both European and U.S. markets with efficiency. It was also able to get more funds and partnerships by showing its regulatory readiness, documented to the investors, which led to faster adoption, along with strict safety regulations.
Conclusion
A key component of innovation in the medical device sector is effectively navigating regulatory pathways. Startups can lower compliance risks, guarantee product quality, and hasten market entry by coordinating product development with FDA, EU MDR, and ISO 13485 requirements. Emerging businesses are empowered to transform regulatory obstacles into chances for expansion and credibility through early planning, strong quality management systems, and strategic alliances with regulatory specialists. In the end, regulatory readiness is more than just compliance; in the highly competitive medical device industry, it is a driver of innovation, trust, and long-term success.
